Graph Embedding on Biomedical Networks: methods, applications and evaluations
Abstract
Graph Embedding learning that aims to automatically learn low-dimensional node representations.
We select 11 representative graph embedding methods and conduct a systematic comparison on 3 important biomedical link prediction tasks: drug-disease association (DDA) prediction, drug-drug interaction (DDI) classification, protein-protein interaction (PPI) prediction; and 2 node classification tasks: medical term semantic type classification, protein function prediction.
The recent graph embedding methods achieve competitive performance without using any bilogical features and the learned embeddings can be treated as complementary representations for the biological features.
Introduction
Graphs have been widely used to represent biomedical entites (as nodes) and their relations (as edges).
The goal of graph embedding is to automatically learn a low-dimensional feature representation for each node in the graph. The low-dimensional representations are learned to preserve the structural information of graphs, and thus can be used as features in building machine learning models for various downstream tasks. Figure 1 summarizes the pipeline for applying various graph embedding methods to downstream prediction tasks.
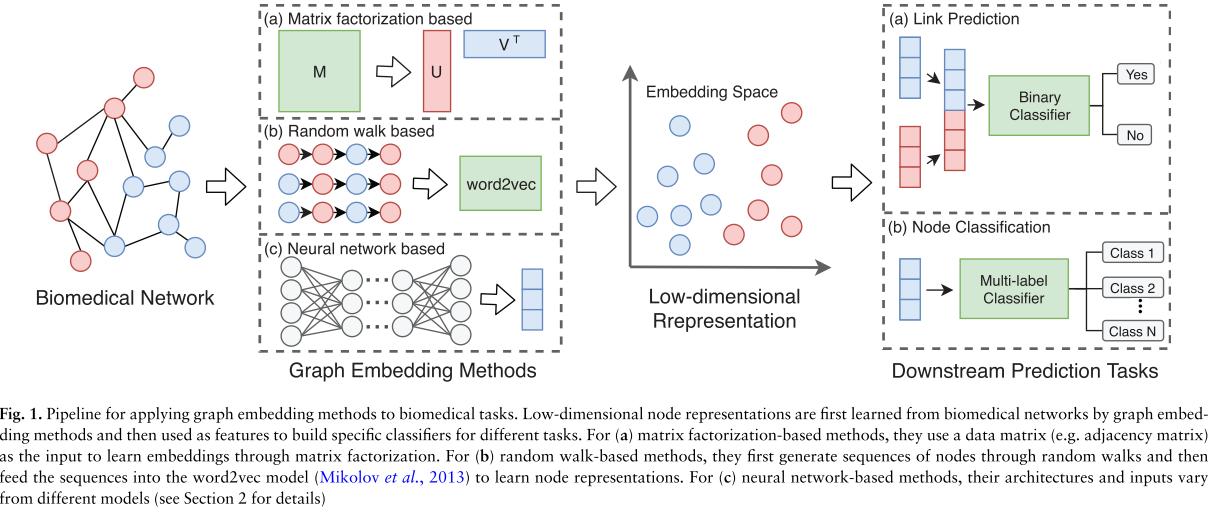
Overview of graph embedding methods
MF-based
Matrix Factorization (MF) has been widely adopted for data analyses. Essentially, it aims to factorize a data matrix into lower dimensional matrices and still keep the manifold structure and topological properties hidden in the original data matrix.
Traditional MF has many variants, such as singular value decomposition (SVD) and graph factorization (GF). And they often focus on factorizing the first-order data matrix.
Random Walk-based
Inspired by the word2vec model, a popular word embedding technique from Natural Language Processing (NLP), which tries to learn word representations from sentences, random walk-based methods are developed to learn node representations by generating node sequences through random walks in graphs. Specifically, given a graph and a starting node, random walk-based methods first select one of the node’s neighbors randomly and then move to this neighbor. This procedure is repeated to obtain node sequences. Then the word2vec model is adopted to learn embeddings based on the generated sequences of nodes. In this way, structural and topological information can be preserved into latent features.
DeepWalk, node2vec, and struc2vec are new work in this category.
Neural Network-based
Various neural networks also have been introduced into graph embedding areas, such as MLP, autoencoder, GAN, and GCN. Different methods adopt different neural architectures and use different kinds of graph infor- mation as input.
- Line directly models node embedding vectors by approximating the first-order proximity and second-order proximity of nodes, which can be seen as a single-layer MLP model.
- DNGR applies the stacked denoising autoencoders on the positive pointwise mutual information matrix to learn deep low-dimensional node embeddings.
- SDNE adopts a deep autoencoder to preserve the second-order proximity by reconstructing the neighborhood strcutre of each node
- GAE utilizes a GCN encoder and an inner product decoder to learn node embeddings.
- GraphGAN adopts GANs to model the connectivity of nodes. The GAN framework includes a generator and a discriminator where the generator approximates the true connectivity distribution over all other nodes and generates fake samples, while the discriminator model detects whether the sampled nodes are from ground truth or genearted by the generator.
Applications of Graph Embedding on Biomedical Networks
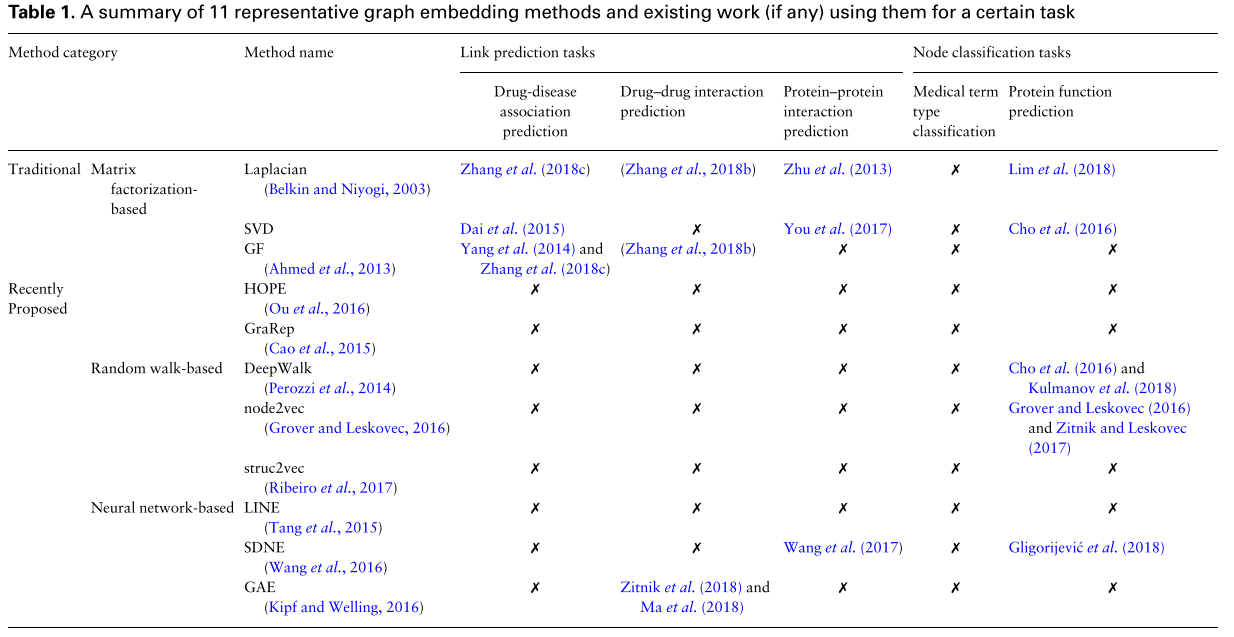
We select 11 representative graph embedding methods and review how they are used on 3 popular biomedical link prediction applications, and 2 biomedical node classification applications.
Link Prediction
Discovering new interactions (links) is one of the most important tasks in the biomedical area.Developing computational methods such as DDA network, DDI network, and PPI network can help generate hypotheses of potential associations or interactions in biological network.
The link prediction task can be formulated as: given a set of biomedical entities and their known interactions, we aim to predict other potential interactions between entities.
Traditional methods in the biomedical field put much effort on feature engineering to develop biological features, gene ontology, or graph properties.
However, deploying methods based on biological features typically faces two problems:
- biological features may not always be available and can be hard and costly to obtain
- One popular approach to solve this problem is to remove those biological entities without features via pre-processing, which usually results in small-scale pruned datasets
- Biological featuers, as well as hand-crafted graph featurs, may not be precise enough to represent or characterize biomedical entities, and may fail to help build a robust and accurate model for many applications.
Graph embedding methods that seek to learn node representations automatically are promising to solve the two problems mentioned above.
- MF-based techniques are used for predictions of DDAs.
- Manifold regularized MF in which Laplacian regularization is incorporated to learn a better drug representations in predicting DDIs.
- PPIs are commonly predicted using Laplacian and SVD techniques.
- Autoencoder-based model to learn embedding of proteins, similar to SDNE
Node Classification
Node classification which aims to predict the class of unlabeled nodes given a partially labeled graph, is also one of the most important applications in graph analysis.
Protein Function Prediction
- A regularized Laplacian kernel-based method is proposed to learn low-dimensional embeddings of proteins.
- Mashup, which first performs random walks with restart on PPI networks and then learns embeddings for each protein via a low rank matrix approximation method.
- DeepGO that learns joint representations of proteins based on protein sequence as well as PPI network.
- OhmNet, which optimizes hierarchical dependency objectives based on node2vec to learn feature representations in multilayer tissue networks for function prediction.
- deepNF, which learns embeddings of proteins via a deep autoencoder.
Medical Term Semantic Type Classification
The increase of clinical texts have been encouraging data-driven models for improving the patient personal care and help clinical decision. To facilitate research on clinical texts, a popular substitute strategy for releasing raw clinical texts, a popular substitue strategy for releasing raw clinical texts is to extract medical terms and their aggregated co-occurrence counts from the clinical texts. However, such datasets do not reveal the semantic information.
A less-investigated but meaning ful node classification task: given a medical term co-occurrence graph where terms and co-occurrence statistics have been extracted from clinical texts, classify the semantic types of medical terms.
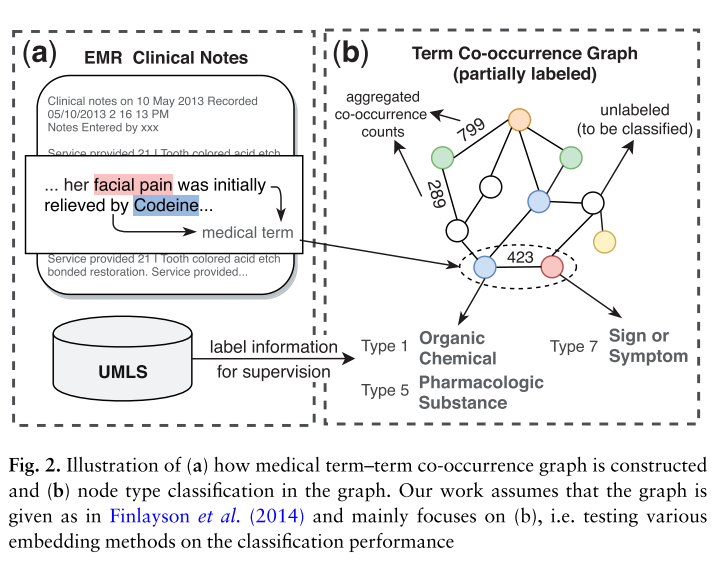
We apply graph embedding methods to the co-occurence graph to learn representations of medical terms. Afterward, a multi-label classifier can be trained based on the learned embeddings to classify the semantic types of medical terms.
Experiments
Datasets
For Link Prediction:
- DDA Graph: Comparative Toxicogenomics Database (CTD). We obtain 92813 edges between 12765 nodes in this graph. Another is National Drug File Reference Terminology (NDF-RT) in UMLS. We extract drug-disease treatment associations using the may treat and may be treated by relationships in NDF-RT. This graph (named ‘NDFRT DDA’) contains 13545 nodes (12 337 drugs and 1208 diseases) and 56515 edges.
- DDI Graph: We collect verified DDIs from DrugBank. We obtain 242 027 DDIs between 2191 drugs and refer to this dataset as ‘DrugBank DDI’.
- PPI Graph: We extract Homo sapiens PPIs from STRING database. Finally, we obtain 359 776 interactions among 15 131 proteins and name this dataset as ‘STRING PPI’.
For Node Classification:
- Medical Term-Term Co-occurrence Graph
- PPI Graphs with Functional Annotations